-
Introduction
This work is an extending development
of our investigations on molecular dynamics simulation of contact melting
[ 1-12]
. Contact melting is a phenomenon of lowering of melting point at the contiguity
zone of the two different crystals. Our molecular dynamics simulations showed
that system of two contact bodies has the new physical properties. It happens
when only masses and sizes of particles are difference in the difference
sides of the boundary. And we are interest by the question: What will as
a result of differences of other system parameters, as desksides of location
of particles, group average velocities, and so on, on sides of section border
of two materials forming the contact? Thus we started the studies of shock
compressed systems [13]
.
The research on shock - compressed conditions
is closely connected with a work on production of amorphous materials, welding
by explosion, making the artificial diamonds, and with other similar works.
Molecular dynamics simulations has been broadly used to study microscopic
features of substances. From the simulation, the time dependence of positions
and velocities of particles are obtained by numerical integrating the equation
of motion. Therefore, the MD method has been used to simulate a nonequilibrium
phenomena. The MD method has been used to simulate crystallisation, glass
formation [14]
and melting processes for many system [15-16]
.
Our studies were conduct for the ionic
system. The alkali halides have been the preferred model system. Their physical
and chemical properties have been widely investigated, and are well understood.
There is a considerable interest in the understanding of phenomena by extreme
conditions, for example, pressure-induced solid-solid phase transition
[17]
.
-
Some shock compression physical phenomena
The feature of processes, which take
place by the shock compression, is a structural modification of surface layers
with practically absent of diffusion. In the Fig.1-2
you can see some simplified scheme of receiving of two aspects of shock
compression phenomena. The welding by explosion is shown in
Fig. 1
. The cumulative effect is shown in Fig. 2
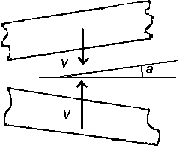
. Beginnings of these processes are alike. Initial location and velocity
of surfaces are shown in Fig 3
. The some combinations of the velocity V and the angle
a are lead to a welding or a cumulation. The cumulative effect
occurs when surfaces have the specific angle. Shock - compression conditions
in the region of the collision of two surfaces gives these effects. It is
known, that welding by explosion can connect two substances, which can not
be connected by another ways. Importance technical significance has phenomenon
of strengthening by explosion showed in Fig 4
.
-
MD simulation of the cumulative effect
The use of simulation on atomic scale
is the possibly best means to finding - out for formation mechanisms under
actions of shock loads and for revealing of processes, which take place thus.
The molecular dynamic method (MD) allows to trace the movement of atoms or
ions under the action of interparticle forces, and also to research the modification
of its structures under actions of shock loads.
We made the simulation for ionic crystals
with the BHM potential for various initial speeds. The estimate of structure
characteristics was implemented by calculation of radial distribution functions
(RDF) and by visualisation of particle trajectories. We note, that the RDF
do not allows to find the great variety of structures, which are formed.
Therefore it is possible to receive the maximal information only by visualisation
of particle trajectories. We made the average of particle co-ordinates for
the time of average in fluctuation period to the exception of influence of
thermal fluctuations on visualisation. The MD-experiments had show the existence
of strong processes of the structural rebuilding and the transfer to the
amorphous structure under shock interaction in surface zone of several crystal
layers. There are observed the cumulative effects on atomic scale of size.
In Fig. 5
you can see the first simplified scheme of molecular dynamic experiment
to study the shock - compression phenomena as the welding by explosion and
the cumulative effect. The initial coordinates and velocities were randomly
varied to provide the best fit to the expected effect.
-
Discussion
In our simulation we have recently discovered
that sometimes certain ions move with more increased velocity and more linearly
can be more long time. It happens by random configuration of particle. We
would like to draw your attention to a question one can raise: Do observed
properties really exist or they are only results of some calculation divergence?
We specially examined situations, when calculation system has calculation
divergence. It may by for example, when started position of two particles
are at a very short, unrealistic distance. The system behaviour is absolutely
different in this case. We think, that received property are true. We are
study the problem.
We can say that there is same computer
evidence for a commulation effect on the scale of hundred particles. With
our small number of computer experiments, it does not seem reasonable to
draw any definite conclusions as the mechanism of commutation. Our statistic
are not rich enough to give conclusive results. Under the microscopic shock
compression lattice becomes excited. We presuppose the stochastic shock influences
are the ordinary events on normal conditions. The external shock action considerable
increases the number of internal overshock events. These overshock have more
stronger specific power. They are the basic reason such phenomena as welding
by explosion and hardening be explosion. Atomic implication of this process
is destruction of old structures and formation the new stable nodes. We should
add, however, that MD simulation is very computer time consuming and our
simulation result should be considered preliminary and our statistic are
not rich enough to give the accurate confirmation of this our ideas.
-
MD simulation of substance properties in
the shock- or unloading-wave
Different way to study the shock-compression
is the simulation of a substance in (a) the significant increasing or (b)
the decreasing of interatomic space. It is the simulation of substance properties
(a) in the shock-wave or (b) in the unloading-wave. Modelling of shock compression
was realised by the changing of the size of the calculation box. For simulation
this effects we use same models. The first model started the calculation
of the ion movements from nodes of the cubical net. Temperature has a broad
range of values. First, we have constructed the initial coordinate and velocity
of ions. Then by use of the numerical methods of temperature stabilisation
we have make the run of MD simulations with different temperatures and values
of size of cubic cell. One initial structure of particle system is regular
cubic lattice of ions as for NaCl, and casual velocities. Second initial
structure is some result of preceding MD simulations. The states of system
have been examined by use of the mean square displacement R2
k(t) of ions
R2k(t
)= <ri(t)-ri(to)}
2>k
(1)
where ri(t)
is position vector of i-th ion at time t, < >
denoted the average, k denoted the type of ions.
We have developed a computer program,
which can be used for simulation of many-bodies problem for classical systems
with central interactions between particles, and can get an evolution in
the time for ensemble of particles. The ensemble consists of 4 ion species,
describing by the corresponding ion positions and velocities. The program
allows one to get the mean-square displacements of ions, diffusion coefficients,
partial radial distribution functions, normalised velocity autocorrelation
functions and another data. Simulation was realised on traditional MD scheme
with using the BHM [18]
and Pauling's potentials. Number of all ions is 216, and the numbers of
each type of ions are 64. Molecular Dynamics Simulation 54 A+
, 54 B-, 54 C+, and 54 D-
were placed in the periodic cube. Our cubic simulation cell is divided initially
by two equal parts corresponding to the AB and CD species respectively.
The time step was 7-10 fs. At the initial stage of the MD simulation, NTV
ensemble was settled. The data analyses were done with the final 1000 steps.
The general statements of numerical experiments are given in table 1. The
AB-CD notation denotes the following: chemical system AB is initially
located in the first half of cubic cell and system BC is initially
located in the second half. The A+,B-,C+
,D- compound denotes an homogeneous ion mixture.
TABLE 1. The general
statements of numerical experiments
|
System composition
|
Initial configuration of ions
|
Some details or/and purpose of experiment
|
|
NaCl-KBr
|
regular ideal cubic lattice
|
an effect of the ion exchange: Cl- and
Br-
|
|
Na+,Br-,K+,Cl
-
|
amorphous state
|
|
|
NaBr-KCl
|
regular ideal cubic lattice
|
|
|
NaBr-KCl
|
regular ideal cubic lattice
|
locations of Br- and Cl-
are fixed
|
|
Na+,Br-,K+,Cl
-
|
amorphous state
|
locations of Br- and Cl-
are fixed
|
The diffusion coefficients Dk
(k=1,2,3,4) have been estimated by using the evolution parameters
of a system and have been calculated by means of the relation
<{ri(t
)-ri(to)}2>k
= 6 Dk t+ Ck , to< t< t,
(2)
The velocity autocorrelation function
is defined as,
Zk(t)=<V
i(0)Vi(t)>k/ <V
i2>k
(3)
where Vi is the velocity
of ions, and k is a number of the ion type.
We have used here two potential approximations by Pauling [7]
and Fumi-Tosi. The Pauling's potential between ions labelled "i"
and "j" is given by
|
Vij (r)= e2/(4
p e
0 r )(qi q j r-1
+ [(si
+sj
)/r] p/(p
+1)
|
(4)
|
where s are
parameters, p = 8 (the hardness parameter),
e0 is the electric
constant, q · e are the ionic charges, e is the electron
charge.
The Fumi-Tosi potential is of the form
Vij(r)= ZiZ
je2/(4 p
e0
r ) + (1+ Zi/ni + Zj/nj) b exp[(
si+
sj-r)/
r]-Cij/r6-Dij/r
8 (5)
here Z is an ionic charge number,
n the number of the electrons in an outer shell, b a repulsion
parameter, s
a value characteristic of an ion size and
r a softness parameter.
- Results